Chapter 5 Suggestions for GSClassifier model developers
5.1 About
The book
R packagesis a straightaway and useful reference book for R developers. The free-access website forR packagesis https://r-pkgs.org/. As a developer of R, if you haven’t heard about it, it’s strongly recommended to just read it. Hadley Wickham, the main author of the book, is an active R developer and has led some masterworks likeggplot2andplyr.With
GSClassifierpackage, it could be easy for users to build a model only with certain gene sets and transcriptomics data. If you are interested in sharing your model,GSClassifieralso provides a simple methodology for this vision. In this section, let’s see how to achieve it!
First, load the package
5.2 Available models
With GSClassifier_Data(), all models supported in the current GSClassifier package would be shown.
GSClassifier_Data()
# Available data:
# Usage example:
# ImmuneSubtype.rds
# PAD.train_20200110.rds
# PAD.train_20220916.rds
# PAD <- readRDS(system.file("extdata", "PAD.train_20200110.rds", package = "GSClassifier"))
# ImmuneSubtype <- readRDS(system.file("extdata", "ImmuneSubtype.rds", package = "GSClassifier"))For more details of GSClassifier_Data(), just:
?GSClassifier_Data()Set model=F, all .rds data would be showed:
GSClassifier_Data(model = F)
# Available data:
# Usage example:
# general-gene-annotation.rds
# ImmuneSubtype.rds
# PAD.train_20200110.rds
# PAD.train_20220916.rds
# testData.rds
# PAD <- readRDS(system.file("extdata", "PAD.train_20200110.rds", package = "GSClassifier"))
# ImmuneSubtype <- readRDS(system.file("extdata", "ImmuneSubtype.rds", package = "GSClassifier"))5.3 Components of a GSClassifier model
Currently, a GSClassifier model and related product environments are designed as a list object. Let’s take PAD.train_20210110(also called PADi) as an example.
This picture shows the components of PADi:
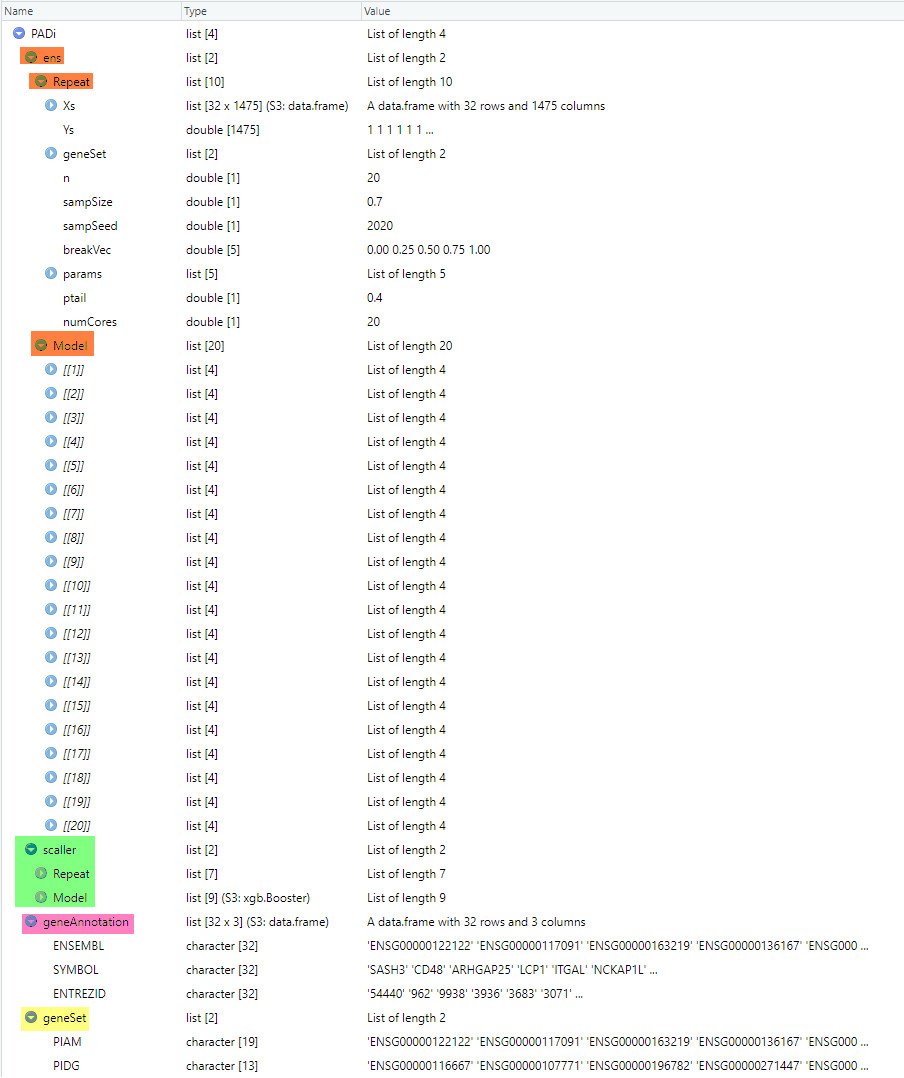
Figure 5.1: Details of a GSClassifier model
As shown, a typical GSClassifier model is consist of four parts (with different colors in the picture):
1. ens:Repeat: productive parameters ofGSClassifiermodelsModel:GSClassifiermodels. Here,PADihad 20 models from different subs of the training cohorts
2. scaller:Repeat: productive parameters of thescallermodel, which was used forBestCallcallingModel: thescallermodel
3. geneAnnotation: a data frame containing gene annotation information4. geneSet: a list contains several gene sets
Thus, you can assemble your model like:
model <- list()
# bootstrap models based on the training cohort
model[['ens']] <- <Your model for subtypes calling>
# Scaller model
model[['scaller']] <- <Your scaller for BestCall calling>
# a data frame contarining gene annotation for IDs convertion
model[['geneAnnotation']] <- <Your gene annotation>
# Your gene sets
model[['geneSet']] <- <Your gene sets>
saveRDS(model, 'your-model.rds')
More tutorials for model establishment, please go to markdown tutorial or html tutorial.
5.4 Submit models to luckyModel package
Considering most users of GSClassifier might not need lots of models, We divided the model storage feature into a new ensemble package called luckyModel. Don’t worry, the usage is very easy!
If you want to submit your model, you should apply for a contributor of luckyModel first. Then, just send the model (.rds) into the inst/extdata/<project> path of luckyModel. After an audit, your branch would be accepted and available for the users.
The name of your model must be the format as follows:
# <project>
GSClassifier
# <creator>_<model>_v<yyyymmdd>:
HWB_PAD_v20211201.rds5.5 Repeatablility of models
For repeatability, you had better submit a .zip or .tar.gz file containing the information of your model. Here are some suggestions:
<creator>_<model>_v<yyyymmdd>.mdDestinations: Why you develop the model
Design: The evidence for gene sigatures, et al
Data sources: The data for model training and validating, et al
Applications: Where to use your model
Limintations: Limitation or improvement direction of your model
<creator>_<model>_v<yyyymmdd>.R: The code you used for model training and validating.Data-of-<creator>_<model>_v<yyyymmdd>.rds(Optional): Due to huge size of omics data, it’s OK for you not to submit the raw data.
Welcome your contributions!
5.6 Gene Annotation
For convenience, we provided a general gene annotation dataset for different genomics:
gga <- readRDS(system.file("extdata", "general-gene-annotation.rds", package = "GSClassifier"))
names(gga)
# [1] "hg38" "hg19" "mm10"I believe they’re enough for routine medicine studies.
Here, take a look at hg38:
hg38 <- gga$hg38
head(hg38)
# ENSEMBL SYMBOL ENTREZID
# 1 ENSG00000223972 DDX11L1 100287102
# 3 ENSG00000227232 WASH7P <NA>
# 4 ENSG00000278267 MIR6859-1 102466751
# 5 ENSG00000243485 RP11-34P13.3 <NA>
# 6 ENSG00000284332 MIR1302-2 100302278
# 7 ENSG00000237613 FAM138A 645520With this kind of data, it’s simple to customize your own gene annotation (take PADi as examples):
tGene <- as.character(unlist(PADi$geneSet))
geneAnnotation <- hg38[hg38$ENSEMBL %in% tGene, ]
dim(geneAnnotation)
# [1] 32 3Have a check:
head(geneAnnotation)
# ENSEMBL SYMBOL ENTREZID
# 353 ENSG00000171608 PIK3CD 5293
# 1169 ENSG00000134686 PHC2 1912
# 2892 ENSG00000134247 PTGFRN 5738
# 3855 ENSG00000117090 SLAMF1 6504
# 3858 ENSG00000117091 CD48 962
# 4043 ENSG00000198821 CD247 919This geneAnnotation could be the model[['geneAnnotation']].
Also, we use a function called convert to do gene ID convertion.
luckyBase::convert(c('GAPDH','TP53'), 'SYMBOL', 'ENSEMBL', hg38)
# [1] "ENSG00000111640" "ENSG00000141510"Note: the luckyBase package integrates lots of useful tiny functions, you could explore it sometimes.